BioVie Inc. today announced positive analysis of unblinded, topline efficacy data from its Phase 3 clinical trial (NCT04669028) of NE3107 in the treatment of mild to moderate Alzheimer’s Disease (AD).
Positive Trending Data from 57 Per-Protocol Patients Suggest NE3107 is Biologically Active and May Have Impact on Cognitive, Functional, and Biomarker Endpoints
Sponsor Identified Issues Relating to Significant GCP Violations and Protocol Deviations, Which Allowed for Data from Only a Subset of Enrolled Patients to be Included in the Efficacy Analysis; Sites Suspected of Improprieties Have Been Referred to FDA
Due to Exclusions, the Primary Efficacy Endpoint Missed Statistical Significance; Adaptive Feature of Trial May Allow the Company to Continue Enrolling Patients to Reach Statistical Significance for Registrational Purposes
Management to Host Conference Call at 8:30 AM ET Today to Discuss Data
CARSON CITY, Nev., Nov. 29, 2023 (GLOBE NEWSWIRE) -- BioVie Inc., (NASDAQ: BIVI) (“BioVie” or the “Company”) a clinical-stage company developing innovative drug therapies for the treatment of neurological and neurodegenerative disorders and advanced liver disease, today announced positive analysis of unblinded, topline efficacy data from its Phase 3 clinical trial (NCT04669028) of NE3107 in the treatment of mild to moderate Alzheimer’s Disease (AD).
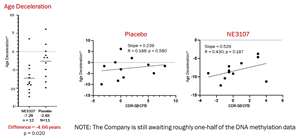
Data from evaluable patients show NE3107’s treatment advantage compared to placebo may be equal to or greater than the benefit from approved AD monoclonal antibodies. NE3107-treated patients also experienced a 4.66-year advantage in age deceleration vs. placebo as measured by epigenetics/DNA methylation Skin Blood Clock.
The trial started during the COVID-19 pandemic when access to clinical sites was limited and enrolled a total of 439 patients through 39 sites. Upon trial completion, the Company found significant deviation from protocol and Good Clinical Practice (GCP) violations at 15 sites (virtually all of which were from one geographic area). This highly unusual level of suspected improprieties led the Company to exclude all patients from these sites and to refer them to the U.S. Food and Drug Administration (FDA) Office of Scientific Investigations (OSI) for further action. After these exclusions, 81 patients remained in our Modified Intent to Treat (MITT) population, 57 of whom were in the Per-Protocol population which included those who completed the trial and were verified to take study drug from pharmacokinetic (PK) data.
“These data show NE3107’s treatment advantage over placebo to potentially be equal to or greater than data reported from clinical trials for the approved medications for AD without the associated safety concerns,” said Cuong Do, BioVie’s President and CEO. “The adaptive trial design gives us the flexibility to continue patient enrollment in the advancement of this potentially important treatment for AD, and we look forward to discussing our findings of NE3107’s magnitude of therapeutic impact with our potential partners. I am also very proud of the integrity our team displayed in taking immediate action to identify and report the potentially problematic sites to the FDA for independent investigation. Importantly, we recognize that along with the development of new and innovative therapeutics, our foremost responsibility in clinical testing is to protect the rights and well-being of study patients and the integrity of the clinical research process.”
Key Findings
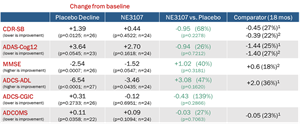
- Patients treated with NE3107 showed improved performance compared to placebo on all cognitive and functional assessments commonly used in the prior approval of amyloid beta (Aβ)-based AD therapies, although the data missed statistically significance due to site exclusions. Placebo-treated patients significantly worsened on virtually all assessments as expected from natural history of the disease. By contrast, NE3107-treated patients experienced a treatment advantage after 6 months that was equal to or greater than results reported from clinical trials for the approved Aβ monoclonal antibody treatments after 18 months.
1 Lecanamab after 18 months; van Dyck et al. N Engl J Med. 2023;388:9-2.
2 Aducunumab after 18 months; Haeberlein et al. J Prev Alz Dis. 2022;2(9):197-210.
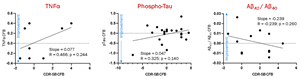
- Improvements in Clinical Dementia Rating-Sum of Boxes (CDR-SB) among NE3107-treated patients appear correlated to changes in tumor necrosis factor alpha (TNFα), plasma phosphorylated tau (p-tau), and the ratio of Aβ42 to Aβ40
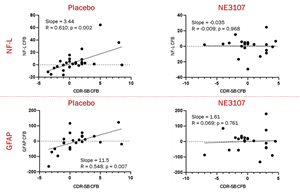
- NE3107 appeared to decrease neuroinflammatory processes that link Neurofilament Light Chain (Nf-L) and Glial fibrillary acidic protein (GFAP), both considered biomarkers of neurodegeneration and cognitive decline.
- Patients treated with NE3107 saw an average of -5.66 years of age deceleration of the DNA methylation advantage compared to those on placebo. Age deceleration is the difference between the patient’s biological age as measured by the Horvath DNA methylation Skin Blood clock less the actual chronological age. NE3107 is believed to be the first drug candidate to demonstrate this impact on DNA methylation and the aging process in a double-blinded, placebo-controlled clinical.
“The unblinded topline efficacy data from 57 per-protocol participants reaffirmed what has been seen in previous studies of NE3107 – which is that patients treated with this molecule appear to experience cognitive and functional improvements as measured by multiple assessment tools,” said Dr. Joseph Palumbo, Executive Vice President, R&D and Chief Medical Officer. “This data reinvigorates our ambition to further evaluate NE3107 and bring the Alzheimer’s community a differentiated treatment that is safe and has a meaningful impact on cognition.”
“The unblinding of topline efficacy data from the trial confirmed an unusual pattern we saw with the blinded data – that patients in a particular demographic group within the trial seemed to have a data pattern different from historical evidence for this demographic group. Patients from this demographic group in this trial reportedly experienced cognitive improvements that were improbable scientifically, and inconsistent with the pathology of this disease,” stated Suzanne Hendrix, Founder & CEO of Pentara, a specialized biostatistics consulting firm that has assisted dozens of AD clinical trials. “When sensitivity analyses were performed, we determined that the anomalous demographic data were associated with the previously identified anomalous sites located in the same geographic area.”
Conference Call & Webcast
| Date | November 29, 2023 |
| Time | 8:30am Eastern Time |
| Conference Call Details | 1-877-407-3982 - Investors Dial (US) 1-201-493-6780 - Investors Dial (Ex US) 13742846 - Conference ID# |
| Webcast Link | Click Here |
| Call Me™ Link | Click Here |
The NCT04669028 Trial – Background & Corrective Actions
The NCT04669028 trial is a Phase 3, double-blind, randomized, placebo-controlled, parallel group, multicenter study of NE3107 in patients who have probable mild- to moderate-AD withs cores on CDR 1-2 and MMSE 14-24. The study has co-primary endpoints looking at cognition using the Alzheimer’s Disease Assessment Scale-Cognitive Scale (ADAS-Cog 12) and function using the Clinical Dementia Rating-Sum of Boxes (CDR-SB). Patients went through two weeks each of 5 mg and 10 mg BID dose titration followed by 26 weeks of 20 mg twice daily vs. placebo, randomized 1:1.
Enrollment
The trial started enrolling patients in August 2021 when COVID-19 pandemic restrictions provided limited access to clinical sites, and the last patient’s last treatment visit was completed in September 2023. A total of 439 patients were enrolled in the trial. BioVie monitored blinded data over the course of the trial to track safety and ensure timely entry of data from the clinical sites into the Electronic Data Capture (EDC, the official database that is submitted to the FDA for registrational purposes) by the Company’s Clinical Research Organization (CRO). The CRO manages the EDC, and each clinical site can enter data only from its patients while BioVie has read-only access to review blinded data.
Data Review and Audit
The Company retained three independent biostatistical consulting firms to analyze the data, including Pentara, which has helped pharmaceutical companies assess dozens of AD trials. Pentara reviewed the blinded data when enough patients completed the trial and identified: 1) several sites in a single geographic area were anomalous (inconsistent data patterns compared to historical data, large proportions of patients improving compared to baseline, unusual data variability); and 2) all patients in a particular demographic group enrolled in this trial seemed to have a data pattern not explainable based on established disease progression and which substantially deviated from historical data for this demographic in other AD trials. Without unblinding and PK data, there was no way to identify the cause of the pattern. Thus, Pentara recommended a subgroup analysis of the identified demographic group vs. all others and anomalous sites vs. others when the study becomes unblinded.
In parallel, some sites started to complete their patient-facing activities in early summer 2023, which created the first opportunity for BioVie to start the data verification and assessment process. The process surfaced unusual data patterns and deviations from expectations (missing data, suspected copied/pasted MRI results, etc.), which led the Company to retain two new CROs to conduct a multi-step process that entailed quality control (QC) visits at all sites, performing source data verification (SDV) on 100% of the documents used in the clinical sites to ensure what was notated on paper during patient visits was accurately reflected in the EDC, and auditing the sites. This extensive, multi-month process concluded when the Company received the final report identifying six sites that appeared to have a large number of deviations from the study protocol and Good Clinical Practices (GCP).
Corrective Actions
Based on the reported findings, and to act responsibly with an abundance of caution, the Company undertook the following before the EDC containing cognitive and functional assessments from the clinical sites was frozen:
- Modified the trial’s Statistical Analysis Plan (SAP) to exclude the 6 sites with GCP and/or protocol deviation from the analysis;
- Amended the study protocol to finalize the primary endpoints to be CDR-SB and ADAS-Cog121 based on our interactions with the FDA and to allow for us to work with FDA to employ the “adaptive design” feature of the trial whereby we might be able to continue enrolling additional patients into the trial should we miss statistical significance due to exclusion of patients from the affected sites; and
- Notified the FDA’s Office of Scientific Investigation (OSI) of the potential issues at the multiple sites with GCP and/or protocol deviation.
As the top-line efficacy data was unblinded and PK data became available, the pre-specified demographic subgroup analyses showed that patients in the identified demographic on placebo significantly improved cognitively without any intervention – a finding that cannot be explained scientifically. Furthermore, the pre-specified anomalous sites vs. others revealed a similar scientifically improbable and that these 9 sites are in the same single geographic area. It turned out that virtually all of the patients in the identified demographic group were associated with the 9 anomalous sites. Consistent with our pre-specified statistical plan, these 9 additional sites were also excluded to arrive at our Modified Intent to Treat population, which became underpowered with just 81 subjects. Out of an abundance of caution, we also referred these 9 additional sites to the FDA’s OSI. It should be noted that virtually all of the 15 sites referred to the FDA were in the same geographic area.
Adaptive Design & Next Steps
The trial was originally designed to be 80% powered with 125 patients in each of the treatment and placebo arms. The unplanned exclusion of so many patients has left the trial unpowered for the primary endpoints. Based on the efficacy signal seen in this trial, the Company intends to work with the FDA to potentially employ the adaptive trial feature of the protocol to continue enrolling patients to achieve statistical significance. The Company has retained a new CRO for future trials.
About NE3107
NE3107 is an oral, small molecule, blood-brain permeable anti-inflammatory insulin sensitizer that binds extracellular signal-regulated kinase. BioVie’s Phase 3 trial is the largest study to date to evaluate the safety and efficacy of NE3107 in patients with AD. NE3107 is the only anti-inflammatory agent currently in phase 3 development for AD. Consistent with the proposed anti-inflammatory and insulin-sensitizing properties of NE3107, this phase 3 study was designed to confirm the efficacy and safety of NE3107 treatment in patients with probable AD.
About BioVie
BioVie Inc. (NASDAQ: BIVI) is a clinical-stage company developing innovative drug therapies for the treatment of neurological and neurodegenerative disorders and advanced liver disease. In neurodegenerative disease, the Company’s drug candidate NE3107 inhibits inflammatory activation of ERK and NFkB (e.g., TNF signaling) that leads to neuroinflammation and insulin resistance, but not their homeostatic functions (e.g., insulin signaling and neuron growth and survival). Both are drivers of Alzheimer’s and Parkinson’s diseases. The Company is conducting a Phase 3 randomized, double-blind, placebo-controlled, parallel-group, multicenter study to evaluate NE3107 in patients who have mild to moderate Alzheimer's disease (NCT04669028). Results of a Phase 2 investigator-initiated trial (NCT05227820) showing NE3107-treated patients experienced improved cognition and biomarker levels were presented at the Clinical Trial in Alzheimer’s Disease (CTAD) annual conference in December 2022. An estimated six million Americans suffer from Alzheimer’s. A Phase 2 study of NE3107 in Parkinson’s disease (NCT05083260) has completed, and data presented at the International Conference on Alzheimer's and Parkinson's Disease and Related Neurological Disorders conference in Gothenburg, Sweden in March 2023 showed significant improvements in “morning on” symptoms and clinically meaningful improvement in motor control in patients treated with a combination of NE3107 and levodopa vs. patients treated with levodopa alone, and no drug-related adverse events. In liver disease, the Company’s Orphan drug candidate BIV201 (continuous infusion terlipressin), with FDA Fast Track status, is being evaluated and discussed with guidance received from the FDA regarding the design of Phase 3 clinical testing of BIV201 for the treatment of ascites due to chronic liver cirrhosis. The active agent is approved in the U.S. and in about 40 countries for related complications of advanced liver cirrhosis. For more information, visit http://www.bioviepharma.com/.
Forward-Looking Statements
This press release contains forward-looking statements, which may be identified by words such as "expect," "look forward to," "anticipate" "intend," "plan," "believe," "seek," "estimate," "will," "project" or words of similar meaning. Although BioVie Inc. believes such forward-looking statements are based on reasonable assumptions, it can give no assurance that its expectations will be attained. Actual results may vary materially from those expressed or implied by the statements herein due to the risk that unblinded data is not consistent with blinded data, the Company's ability to successfully raise sufficient capital on reasonable terms or at all, available cash on hand and contractual and statutory limitations that could impair our ability to pay future dividends, our ability to complete our pre-clinical or clinical studies and to obtain approval for our product candidates, our ability to successfully defend potential future litigation, changes in local or national economic conditions as well as various additional risks, many of which are now unknown and generally out of the Company's control, and which are detailed from time to time in reports filed by the Company with the SEC, including quarterly reports on Form 10-Q, reports on Form 8-K and annual reports on Form 10-K. BioVie Inc. does not undertake any duty to update any statements contained herein (including any forward-looking statements), except as required by law.
For Investor Relations Inquiries:
Contact:
Bruce Mackle
Managing Director, LifeSci Advisors, LLC
bmackle@lifesciadvisors.com
For Media Relations Inquiries:
Melyssa Weible
Managing Partner, Elixir Health Public Relations
mweible@elixirhealthpr.com
1 CDR-SB = Clinical Dementia Rating-Sum of Boxes; ADAS-Cog12 = Alzheimer’s Disease Assessment Scale-Cognitive
Photos accompanying this announcement are available at
https://www.globenewswire.com/NewsRoom/AttachmentNg/5be903ff-7666-42f7-aa2d-f5848763253f
https://www.globenewswire.com/NewsRoom/AttachmentNg/70cd2268-3bb2-4644-81e7-0630d04590d6
https://www.globenewswire.com/NewsRoom/AttachmentNg/539d1325-cc25-4bbb-a394-eebddbde640f
https://www.globenewswire.com/NewsRoom/AttachmentNg/3f25bcdf-6ab6-4745-ae43-f55ff96793b3

![]()
Table 1
Change from Baseline
Figure 2
Improvements in Clinical Dementia Rating-Sum of Boxes
Figure 3
Decrease of Neuroinflammatory Processes
Figure 4