
InfuTronix, LLC has announced a voluntary recall of the Nimbus Ambulatory Infusion Pump System, including Nimbus II PainPro, Nimbus II Flex, Nimbus II Plus, Nimbus II EpiD and Nimbus II EMS from the US Market due to a high number (3698) of customer complaints related to the Nimbus Infusion Pump systems dated May 2019 to August 2023.
NATICK, MA / ACCESSWIRE / March 20, 2024 / InfuTronix, LLC has announced a voluntary recall of the Nimbus Ambulatory Infusion Pump System, including Nimbus II PainPro, Nimbus II Flex, Nimbus II Plus, Nimbus II EpiD and Nimbus II EMS from the US Market due to a high number (3698) of customer complaints related to the Nimbus Infusion Pump systems dated May 2019 to August 2023. Evaluation of complaint data has identified several potential product issues:
Nimbus Pump System
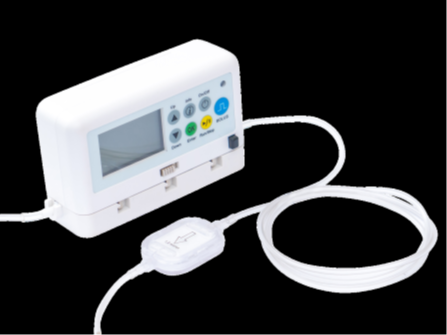
- Battery Power may potentially affect the performance of the pump by causing an immediate power off event.
- Upstream Occlusion, as noted by the upstream occlusion alarm, occurs when there is a block in flow of the proximal end of the administration set.
- System Errors, as noted by the System Error alarm which causes the pump to suspend the infusion.
- Drug product egress from certain administration set bonding points, which may potentially result in drug product leaking from the device.
- Flow Rate (high or low) which may potentially lead to the pump infusing an inaccurate delivery of the drug.
- Pump Housing design which may potentially result in damage over time to certain areas of the housing responsible for administration set engagement, leading to false occlusions and flow rate inaccuracies.
These product issues were identified through the InfuTronix post-market surveillance system and evaluated through the InfuTronix Corrective Action/Preventive Action (CAPA) system. InfuTronix has determined that the best corrective and preventive action to address the identified product issues and potential outcomes is a redesign of the Nimbus Infusion Pump system. The redesign of the system will allow InfuTronix to improve several aspects of the product including mechanics, electronics, software, and housing design as well as aspects of the administration set. Given the number of anticipated design improvements and the extensive requirement for design, verification, and validation, InfuTronix believes a new premarket notification(s) and clearance from FDA may be required. As such, InfuTronix is seeking to remove the system from the market while these improvements and design changes are being made and a new clearance(s) is obtained.
A health hazard evaluation (HHE) was performed by an independent physician to evaluate routine post-market complaint failure modes associated with the Nimbus family of infusion pumps. The HHE determined that the common device failure modes pose a low risk to users.
The Nimbus Infusion Pump system has been distributed throughout the United States since October 17, 2014 until February 21, 2024. It has never been distributed internationally.
Affected devices have the following Unique Device Identification numbers associated with them:
- Nimbus Ambulatory Infusion Pump 00817170020000
- Nimbus II PainPRO 00817170020086
- Nimbus II Flex 00817170020093
- Nimbus II Plus 00817170020161
- Nimbus II EpiD 00817170020376
- Nimbus II EMS 00817170020109
Users may continue to use the Nimbus Infusion Pump system and associated infusion sets during this removal process. Users should be aware of the signs indicating a potential issue with the pump (How to recognize that a device may fail):
- Battery - The infusion pump will brown out (fail-safe). The user will see that the pump has shut off. Additionally, the LED screen may flicker prior to failure, indicating a potential battery issue.
- Upstream Occlusion - Auditory and visual occlusion alarm alerts the user of the occlusion.
- System Error - Auditory and visual system error alarm will alert the user.
- Drug Product Egress - The user may notice leaking drug from the pump pouch or infusion set or feel wetness from the drug product.
- Flow Rate (high/low) - Auditory and visual occlusion alarm for low flow will alert the user of occlusion. The user will notice residual volume remaining at the end of therapy. High flow rate may be associated with shortened infusion times.
- Pump Housing Damage - The user may notice broken latches, hinges, or cracks in the housing and/or user may notice that the device is difficult to assemble prior to use.
Infutronix has already notified customers with a letter that details the products impacted, reason for the voluntary removal, risk to health assessment, how to recognize the device may fail and actions to be taken by the customer/user.
The continued use of the Nimbus Infusion Pump system and associated infusion sets is safe during this removal process. Users should be aware of the signs indicating a potential issue with the pump as described above, "How to recognize that a device may fail".
As with any use, health care providers (HCPs) should read the Instructions for Use (IFU) and follow those closely to ensure proper performance of the pump with its associated accessory set. Further, HCPs should educate patients on proper use of the pump and accessories, and potential operational concerns.
As a reminder to HCPs,
- When replacing batteries, only new batteries should be used. Care should be taken to ensure that old and new batteries are not confused when completing the replacement. When resetting the battery state, HCPs are encouraged to pay close attention to the device indicator lights and LED screen indicating battery life.
- HCPs should take care to clear an occlusion alarm properly. The battery should not be cycled (turned on/off) to clear the occlusion alarm. If the occlusion alarm cannot be cleared, a new pump should be used. Patients should be educated that silencing the alarm does not clear the occlusion.
- Due to the potential ambulatory use of the pump, patients may carry the drug product dispensing pouch in a carrying pack. Patients should be educated that impeding the tubing set or placing pressure on the device (leaning against it, sitting on it) may cause the device to malfunction.
- HCPs are encouraged to educate patients on potential drug leakage and establish appropriate containment protocols for users to follow.
HCPs should ensure users are educated to recognize audio and visual alarms and inspect their device for damage. Any difficulties or abnormalities noted with the pump or accessories should result in immediate discontinued use of the pump and notification to the HCP.
Customers of InfuTronix who have Nimbus Infusion Pumps and associated infusion sets in their possession should contact InfuTronix customer service at Customerservice@intuvie.com, where they will be instructed on the Return Material Authorization (RMA) process to be followed for returning both the Nimbus Infusion Pumps and associated infusion sets. Customers who purchase the Nimbus Infusion Pump and/or associated infusion sets from a distributor should contact their distributor directly and receive instructions on the RMA process to be followed.
As the device will not be available or supported after June 20, 2024, InfuTronix encourages the healthcare provider to seek alternative methods of drug product infusion appropriate for their patient's needs, based on their medical expertise, at the earliest possible opportunity.
InfuTronix is removing the Nimbus Infusion Pump Systems from the market. The products will no longer be supported by InfuTronix for either Nimbus Infusion Pumps or related infusion sets beyond June 20, 2024. To return Nimbus products customers should follow the instructions provided in the Medical Device Removal letter that was sent by InfuTronix, contact InfuTronix customer service or their local distributor.
For further inquiries, please contact InfuTronix Customerservice@intuvie.com or call 508-650-2008, Option 8, Monday through Friday between the hours of 9:30AM and 5:00PM, Eastern Time (ET).
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA's MedWatch Adverse Event Reporting program either online, by regular mail or by fax.
- Complete and submit the report Online: www.fda.gov/medwatch/report.htm
- Regular Mail or Fax: Download form www.fda.gov/MedWatch/getforms.htm or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
SOURCE: InfuTronix
View the original press release on accesswire.com



